 3G Eden
3G Eden RSS
RSSBlue Gene®/L 超级计算机为科学家提供了一流的计算能力和复杂的数据虚拟化工具,帮助他们深入学科的前沿。了解此项技术如何帮助计算分子生物学家创建蛋白质折叠和错误折叠模拟,以更多好理解这些复杂的分子。
2001 年,IBM 的研究科学家着手开始设计一种新的服务器系列,即现在的 IBM System Blue Gene®。这些服务器从 2004 年开始投入使用 — 首先是 Blue Gene/L(本文所讨论的主题),然后是 Blue Gene®/P。
Blue Gene 系列超级计算机采用标准的编程环境,旨在提供超大规模的性能;它们还旨在提高电能、散热和厂房的效率。许多大学、政府和商业研究实验室都使用 Blue Gene 研究射电天文学、蛋白质折叠、气候、宇宙学和药物开发。该系统为科研方式带来的变化是相当巨大的,因为它提供了一个更加高效的工具,用于设计和运行替换版本的复杂模型。
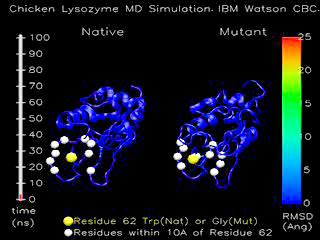
在本文中,我们将展示与蛋白质折叠相关的某个项目的研究进展情况。图 1 显示了我们当前可以完成的工作规模,这得益于 Blue Gene/L 的强大功能。初始配置从溶解酵素晶体结构开始(见 参考资料)。
图 1. 活细胞 10 微秒活动情况;观看视频
蛋白质组学:蛋白质经济
蛋白质是一种生物高分子,它是有机体的基本成分,并且参与了细胞中的每一个流程。许多蛋白质是催化生物反应的酶;一些涉及细胞信号和免疫响应;还有一些提供了肌肉和骨骼的结构和机械功能。演示蛋白质普遍性和重要性的两个例子包括:
- 一种蛋白质负责血液的 “红色状态”;它从肺部携带氧气到身体的所有其他部分。
- 另一种蛋白质负责人类身体对毒葛毒素的响应;刺激性极强,并通常无害。
参与地球上生命活动的蛋白质达到成千上万种。蛋白质组学的研究对象是蛋白质的工作原理、交互方式以及它们的多样化和专一性在生命有机体中的发展演变。本文将简要讨论蛋白质的概念,它们的成分以及它们对所在系统的影响。
DNA 是每一个植物和生物细胞中的信息存储成分。它将信息存储为化学基本成分序列(核苷),我们将其称作 A、C、T 和 G(表示 DNA 中的腺嘌呤、胞嘧啶、胸腺嘧啶和鸟嘌呤,以及 RNA 中的尿嘧啶代替胸腺嘧啶)。从远处看,这些基本成分看上去极为相似,因此 DNA 的每一部分的基本形状都是相同的 — 著名的沃森克里克双螺旋结构。
为了读取 DNA 中的信息,DNA 螺旋分解和另一种 RNA 分子将由内部模式表示构成。您看到的不是拆分结构,而是整体的组合效果。RNA 分子在一旁表示为核糖体的蓝图,这是一种功能类似于全能工厂的蛋白质。核糖体以三个一组的形式读取 A/C/T/G 编码,这样我们可以得出一个 64 字母的 “字母表”。
其中的 12 个 “字母” 对应于氨基酸,它是蛋白质的基本成分。这些氨基酸主要来自我们吃的食物(人不能合成所需的所有氨基酸,因此改组从食物中获取其他的 “基本” 氨基酸)。每种氨基酸都有一个 “头部” 和一个 “尾巴”。核糖体找到每个 “字母” 的适当的氨基酸,并将它们首尾相连,组成一个序列;其他 “字母” 表示何时开始以及何时停止。最终生成的氨基酸线性序列是最新生成的蛋白质分子,它是根据所使用的 DNA 部分中的代码精确形成的。
蛋白质分子各原子之间的压力和张力、与细胞中盐水之间的细微交互以及热量 的随机变化,造成蛋白质分子 “折叠” 成典型的形状。
蛋白质分子相当稳定;其中一些可以持续存在数百年时间而保持不变,以及承受几百摄氏度的高温,这个温度会杀死由它们构成的有机体。它们基本上会保持自身的状态,直到由强烈的化学物质、高压、热或冷改变特性,或者成为其他生物的食物。
它们的形状以及随时间、温度、和周围分子的变化将决定蛋白质分子的行为 — 无论是运输氧气,带来毒素敏感症状,或者任何其他细微的工作。
图 2 展示了熟悉的 DNA 球棍模型(图像提供了一对立体模型;见 参考资料):
图 2. DNA 球棍模型
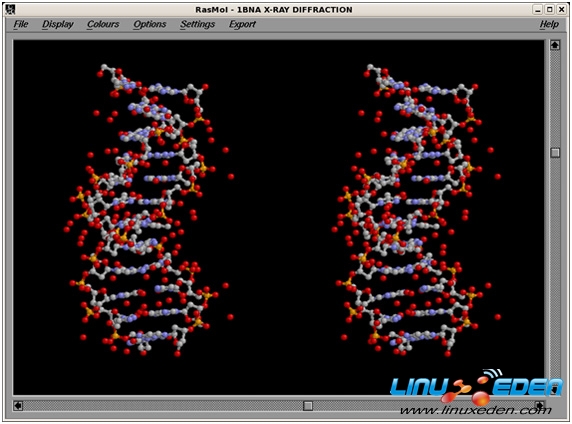
图 3 显示了色氨酸,二十种标准氨基酸中的一种(图像提供了一对立体模型;见 参考资料)。
图 3. 色氨酸,二十种标准氨基酸中的一种
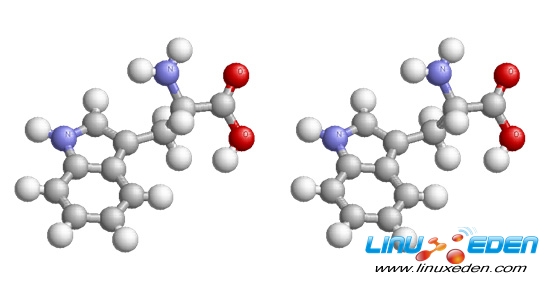
氨基酸合成蛋白质的方式是切断一个分子的 O-H 组(图 3 右侧),切断另一个分子 H 与 N 的连接(图 3 顶部),并将分子拼合起来。所有氨基酸都具有这种特有的原子分组特性(图 3 右侧)。
图 4 显示了蛋白质血色素的视图(图像提供了一对立体模型;见 参考资料)。
图 4. 蛋白质血色素
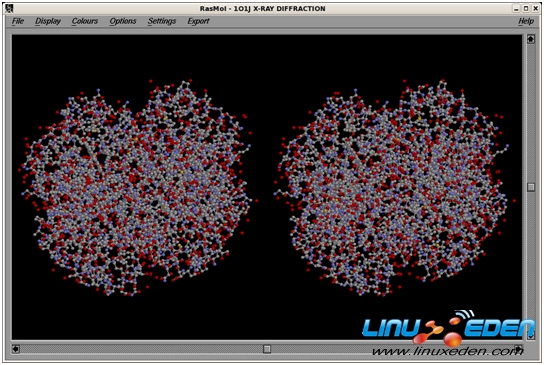
血色素由 4 个子组中的 574 个氨基酸分子组成。血色素,及其相关铁原子(它们合成蛋白质的方式不在本文讨论范围之内),在血液中运输氧气。氧气运输系统只需铁原子便可运行,但血色素提供的蛋白质 “笼状” 结构能够大大提高其效率。如果您将这个图像放到立体图像查看器中,您可以分辨出 3D 原子结构;对于更加复杂的结构,我们需要采用不同的方式来可视化它们。
商业和学术动机
|
开发医药器和保护公共健康的发展越来越依赖于对于生命基本成分的良好理解,比如蛋白质。当前一个颇为关注的话题是 蛋白质错误折叠和聚合 — 如果某个蛋白质折叠成的形状不是目标形状,则最终可能会生成带有不同属性的非活动蛋白质,这会导致一些神经退行性疾病,比如说阿尔茨海默氏症、亚急性海绵样脑病、牛海绵状脑病(疯牛病)亨廷顿和帕金森、囊肿性纤维化以及其他淀粉样变性病。
理解造成蛋白质分子从有用的折叠形式转变成不同折叠形式的原因是研究这些重要疾病的活跃主题。最后,由 Chris Dobson 及其剑桥大学同事组织的实验(见 参考资料)显示,淀粉和纤维不仅可以形成传统的淀粉样肽,而且给予适当的条件就可以形成几乎所有的蛋白质(如溶菌酶)。事实上,溶解酵素蛋白质的单一变异(W62A)可以造成蛋白质的稳定性低于在野生型(参见侧栏);它还可以造成错误折叠,并在针对缺少重要 “远程恐水症交互” 的尿素解决方案中形成可能的淀粉。
科学目前尚未理解这种单一 W62 剩余物在折叠过程中如何在远程恐水症交互中发挥重要的作用,然后由于机能原因从预测的成核位置切换到表面。这可以让人们更好地理解单突变效果,以及与蛋白质错误折叠及聚合相关的上述疾病的内部机制。
Blue Gene/L 技术提供了一种强大的方法来研究这些类型的疾病,因为它能够更加经济高效地(和更快地)建模蛋白质折叠和错误折叠的效果。
我们的建模对象是什么?
图 1 的来源 视频 呈现了由于单一突变造成的溶解酵素蛋白质的序列的一部分。溶解酵素是一种蛋白质,它是人类免疫系统的一部分;由于功能正常,它可以刺破入侵细菌的细胞壁并毁灭它。
单一突变,DNA 序列发生变化,会造成核糖体在构建溶解酵素分子时使用不同的氨基酸。其理论是,这种不同的氨基酸会影响溶解酵素折叠成的形状,并且形状稍有不同的溶解酵素分子在刺破细菌细胞壁方面的效果也不同。理解这种变化之后,我们可以设计药品或其他形式的治疗方法,来帮助发生这种突变的个人从细菌疾病中恢复。
作为工作的一部分,我们将一个溶解酵素分子中的每个原子的位置和速度,以及大约 10,000 个水和尿素分子的位置和速度(此模拟在 8 种摩尔尿素溶液中进行,以模拟实验环境),存储在计算机的内存中。可以通过许多方式来模拟原子之间的作用力;我们使用一种变化的球和弹簧 模型来模拟互作用力,使用 “逆平方法则” 模型来模拟带电原子之间的静电力,并使用 “吸引/击退” 模型来模拟相互接近但没有共价键的原子。模型作为时间序列运行。在每个步骤中,我们计算每个原子受到的作用力,然后根据牛顿第二法则更新速度和位置。
在每个时间步骤中(非常短,差不多 1 飞秒),需要计算的作用力的数量基本上可以达到数亿。计算如此庞大,并且我们还希望能够模拟足够长的时间(微秒)以便模拟感兴趣的运动,这意味着这种方法只在最近才实际可行。有关我们工作以及一些替代方案的详细信息,请参见 参考资料 中的 “Destruction of long-range interactions by a single mutation in lysozyme” 链接。
配置实验室
在纽约约克镇的 IBM Watson Research Lab,我们搭建了 20 组 BlueGene/L 服务器。每台服务器配备 1,024 PowerPC® 双核微处理器芯片;每个微处理器使用 512MB 内存。对于此计算网格 中的每 64 个芯片,都有一个额外的微处理器与 1Gbps Ethernet 相连。这 320 个 Ethernet 链接通过一个标准的以太交换器连接到带有磁盘、磁带、语言编译器和作业控制软件的标准 IBM Power Systems 机器。
这项溶解酵素建模工作平均要使用 4 组 BlueGene/L 处理器花费几个月的时间来生成超过 10 微秒的分子动态数据。应用程序会定期记录模拟中的所有原子的位置和速度(此信息的一部分用于生成上面提到的 合成视频)。在需要重新启动模拟时,可以加载一组合适的位置和速度。重新启动可能需要在计划关机之后,计划外机器故障之后,或者在使用不同时间步骤粒度重放科学模型事件时。